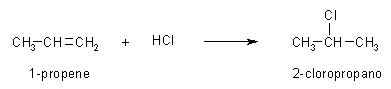|
Pianeta
Chimica.it Responsabile: Prof. Mauro Tonellato |
MODELLISTICA
MOLECOLARE CON ArgusLab
2^ LEZIONE CARBOCATIONE 1° E 2° ALCHENI MONO E DISOSTITUITI |
||
|
Lezioni con
|
Gli argomenti di questa lezione sono: |
||
|
|
|
||
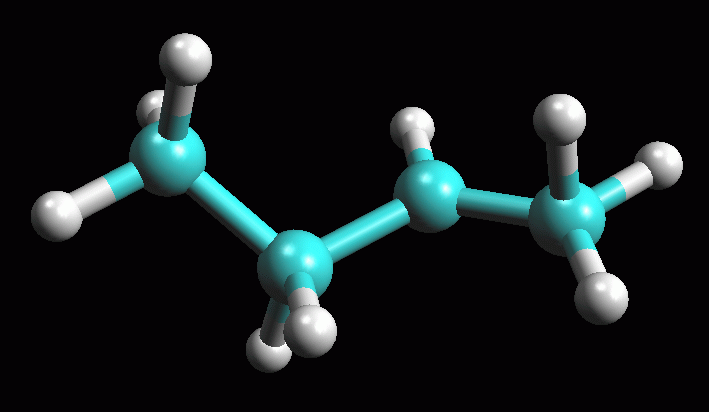 fig. 1 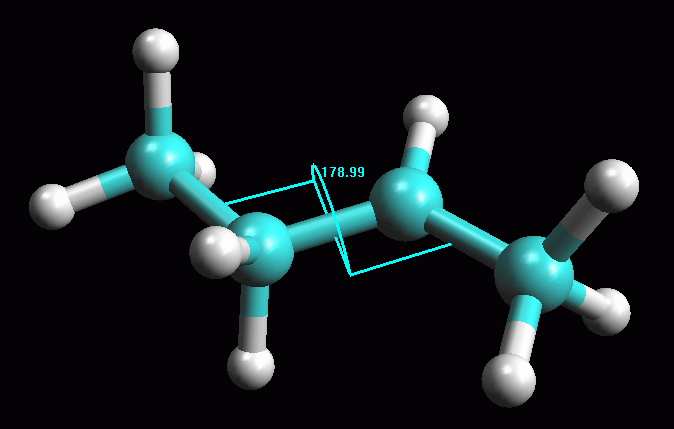
fig. 2  fig. 3 |
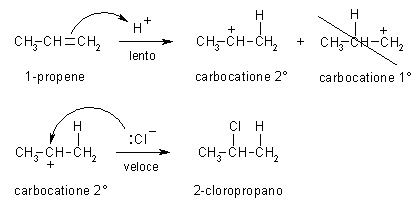
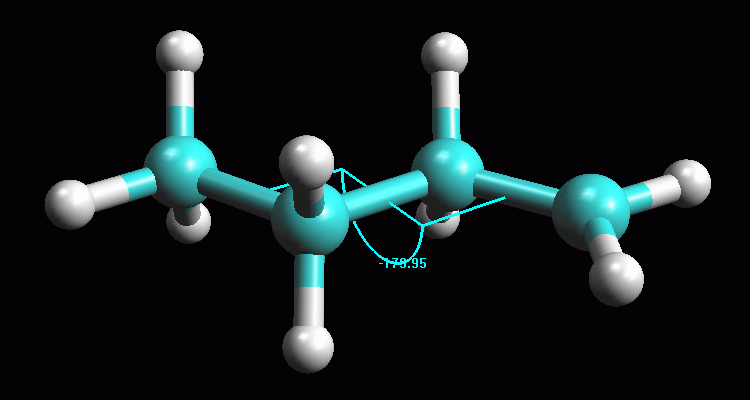
Creare la molecola del carbocatione 2° -- Create la molecola di 1-butene, oppure aprite 1-butene_AM1 salvata durante la prima esercitazione. -- Create il carbocatione 2°. Per simulare l'attacco di H+ a 1-propene mostrato nella figura qui sopra, cambiate il C1, il primo carbonio del doppio legame, da C[sp2] a C[sp3] (Click col pulsante destro, Change Atom) e poi aggiungete l'atomo di idrogeno (pulsante H). Per togliere l'indicazione di doppio legame, fate Click destro sul doppio legame e selezionate Single. Avete ottenuto il carbocatione 2° mostrato in figura 1, quello che si forma quando l'1-butene reagisce con H+ e obbedisce alla regola di Markovnikov. -- Salvate la molecola. Create una nuova directory chiamata Carbocationi e salvate la molecola col nome butil_CC2 (butil CarboCatione secondario). -- Ottimizzate la geometria con UFF e poi AM1. Dopo aver ottimizzato con UFF (più volte), se tentate di ottimizzare la geometria molecolare col metodo AM1 il programma segnala un errore perchè la molecola neutra contiene 25 elettroni quindi è un radicale. Dovete specificare che è un catione e arrivare a 24 elettroni. Nella finestra Geometry Optimization, Molecule portate la carica a 1 (Net Charge). Ottimizzate con AM1, premete Start, otterrete la molecola di fig 2. Controllate che gli idrogeni dei metili siano sfalsati come in figura. Notate che la molecola si è raddrizzata portando l'angolo diedro a 180°. -- Calcolate l'energia della molecola. Calculation, Energy, AM1, Start e poi copiate il valore dell'entalpia di formazione su Word. Dovreste ottenere circa DH = 183,94 kcal/mol (fig 3). -- Salvate come butil_CC2_AM1. |
||
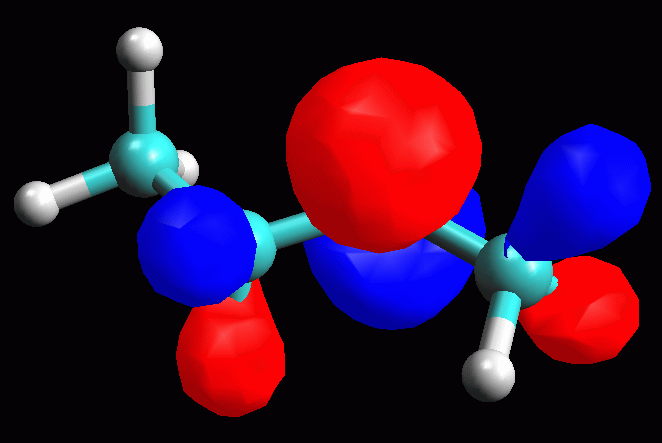 fig. 4 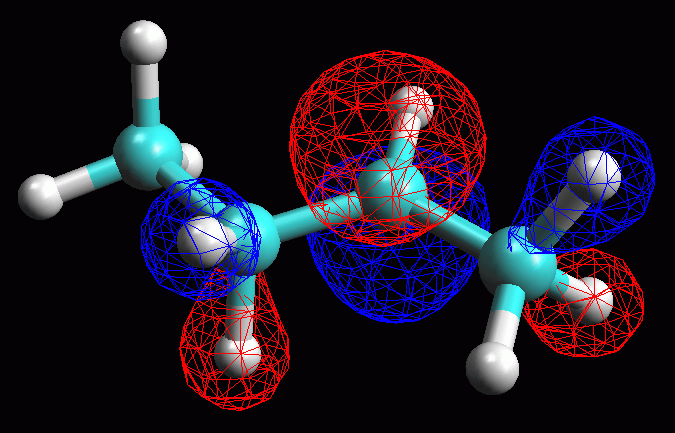
fig. 5 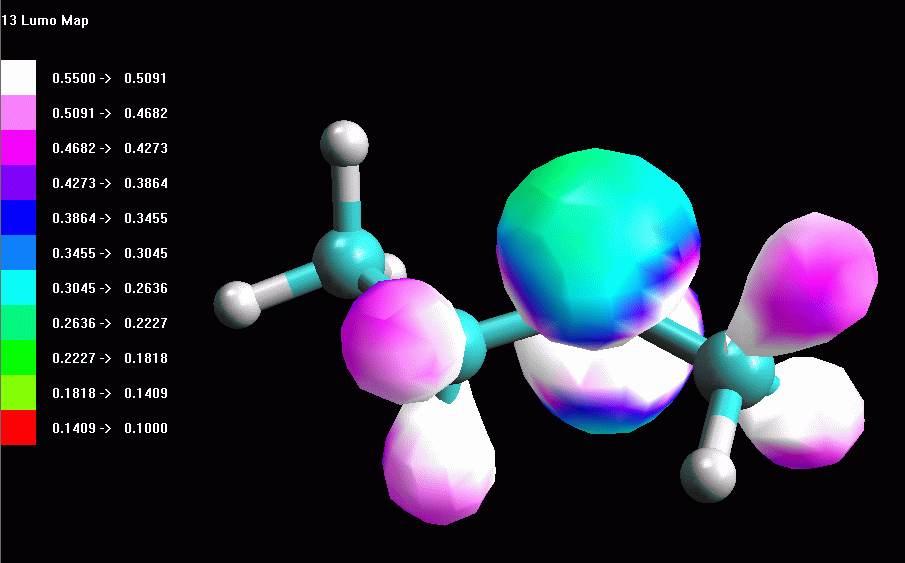 fig. 6 |
Esaminare l'orbitale molecolare LUMO LUMO = orbitale molecolare non occupato a più bassa energia (Lowest Unoccupied Molecular Orbital). LUMO è quindi è l'orbitale vuoto più conveniente per accettare gli elettroni di un nucleofilo che attacca la molecola, in questo caso il Cl- che attacca il carbocatione secondario. La struttura dell'orbitale LUMO ci può aiutare a capire la reattività della molecola verso i nucleofili. Inoltre ci può aiutare a comprendere la diversa stabilità dei carbocationi 1° e 2°. -- Generate l'orbitale LUMO. Scegliete Calculation, Energy, AM1, Surface Properties, 13 LUMO, Start. -- Generate la mappa del potenziale elettrostatico. Calculation, Energy, AM1, Surface Properties, Electrostatic Potential, Start. -- Rappresentate l'orbitale LUMO: Surfaces, Make Surfaces, RF MOs, cubo blu di MO13 trascinato in Grid, scrivete 13 LUMO in Surface Name, Create>>, accendetelo con Toggle Display, OK (fig 4 e 5). Osservando l'orbitale, notate che questo non è localizzato solo sul C2, ma abbraccia anche gli orbitali sp3 che legano gli idrogeni sui carboni vicini (C1 e C3) come suggerisce del resto la teoria dell'iperconiugazione. -- Rappresentate l'orbitale LUMO mappato con il potenziale elettrostatico. Surfaces, Make Surfaces, Mapped, RF MOs MO13 trascinato in Grid 1, Electrostatc Potential Esp 0 trascinato in Grid 2, scrivete 13 LUMO Esp, Create>>, spegnete il vecchio orbitale e accendete il nuovo con Toggle Display, OK. Si ottiene un orbitale tutto bianco, cioè troppo positivo per rientrare nel range mappato che va da -0,05 a +0,05. L'orbitale è anche troppo gonfio quindi fate Click destro - Contour value - 0,05. Ripetete l'operazione di creazione dell'orbitale mappato scegliendo un diverso range di potenziali per esempio da +0,20 a +0,55. (fig 6). Salvate col nome di butil_CC2_Lumo_map. In questa figura si vede molto bene che una parte della carica positiva viene delocalizzata sui lobi laterali. Questo alleggerisce la quantità di carica positiva sul lobo centrale, cioè sul carbocatione. Il carbocatione secondario può delocalizzare un po' della sua carica positiva sui due carboni laterali stabilizzandosi con un meccanismo simile a quello suggerito dalla teoria dell'iperconiugazione, cioè sovrapponendo l'orbitale pigreco centrale con gli orbitali sp3 che legano gli idrogeni sui carboni laterali. |
||
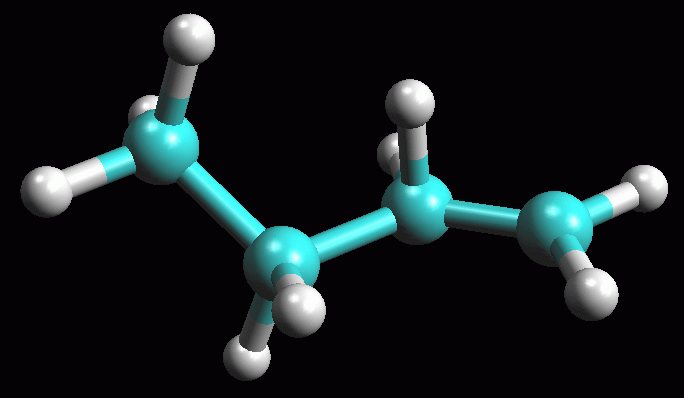 fig. 7
|
Creare la molecola del carbocatione 1° -- Create la molecola di 1-butene, oppure aprite 1-butene_AM1 salvata durante la prima esercitazione. -- Create il carbocatione 1°. Per simulare l'attacco di H+ a 1-propene mostrato nella figura ad inizio pagina, cambiate il C2, il secondo carbonio del doppio legame, da C[sp2] a C[sp3] e poi aggiungete l'atomo di idrogeno (pulsante H). Fate Click destro sul doppio legame e selezionate Single. Avete ottenuto il carbocatione 1° mostrato in figura 7, quello che si formerebbe se 1-butene reagisse con H+ contravvenendo alla regola di Markovnikov e l'H+ si legasse sul secondo carbonio. -- Salvate la molecola col nome butil_CC1 (butil CarboCatione primario). -- Ottimizzate la geometria molecolare col metodo UFF e poi con AM1 (più volte), ma ricordatevi di portare la carica netta a +1. Cliccate Start. Se necessario aiutate manualmente la molecola ad assumere la giusta conformazione con un angolo diedro di 180° come in figura 8 ed ottimizzatela nuovamente. -- Calcolate l'energia della molecola, Calculation, Energy, AM1, Start e poi copiate il valore dell'entalpia di formazione su Word. Dovreste ottenere circa DH = 199,93 Kcal/mol (fig 9). -- Salvate come butil_CC1_AM1. La differenza di entalpia tra i due carbocationi risulta abbastanza grande (183,94 - 199,93 = - 16,00 Kcal/mol). carbocatione 1° => carbocatione 2° ................... DH = - 16,00 Kcal/mol Da qui potete calcolare la loro K di equilibrio applicando la formula: DG° = - RT lnK quindi lnK = - DG° / RT lnK = 16 . 4184 / 8,31 . 298 = 27,03 Usando DH° al posto di DG°, e quindi trascurando l'entropia, otteniamo un valore indicativo per la K di equilibrio che risulta K = 5,48 1011. Da questi dati potete concludere che l'equilibrio tra i due carbocationi è talmente spostato a favore del carbocatione 2° che quello primario praticamente non si forma. La regola di Markovnikov trova quindi ampia giustificazione da questa analisi della stabilità dei due carbocationi. |
||
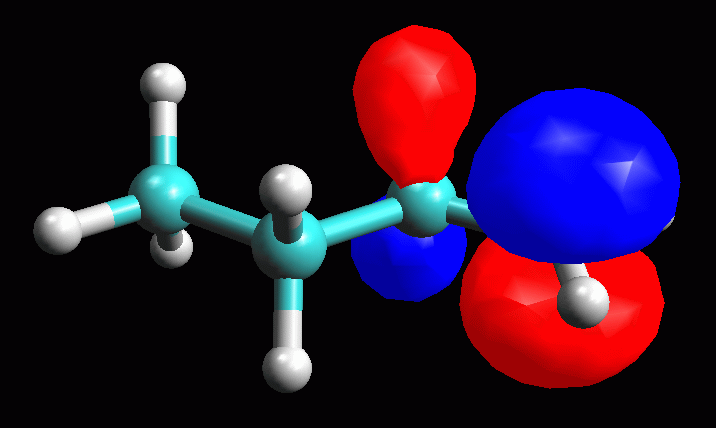 fig. 10 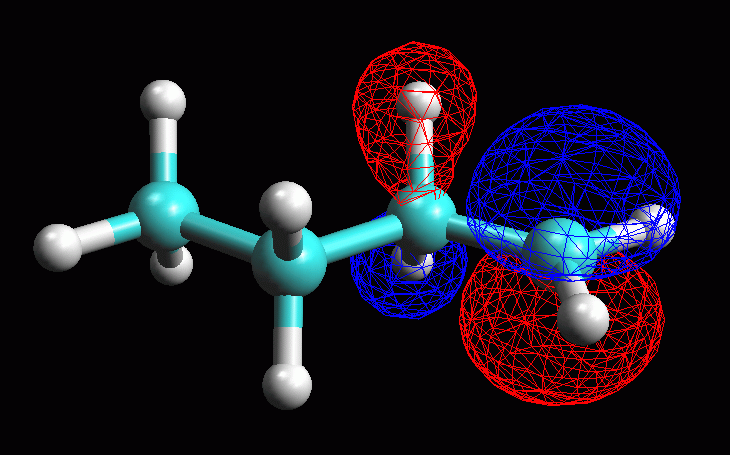
fig. 11 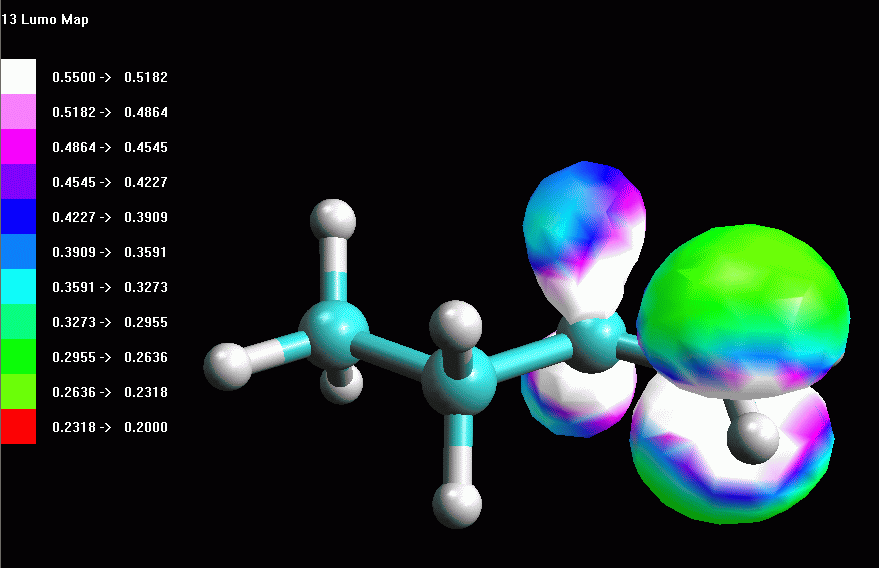 fig. 12 |
Autore: prof Mauro Tonellato |
||
|
|
|
||
|
|
|||