|
Pianeta
Chimica.it
Responsabile:
Prof. Mauro Tonellato
|
|
MODELLISTICA
MOLECOLARE CON ArgusLab
1^ LEZIONE
1-BUTENE E 2-BUTENE
|
|
| |
|
|
|
|
Lezioni con
ArgusLab
 1-butene
e 2-butene 1-butene
e 2-butene
Carbocatione
1° e 2°
Acetone
e tautomeria
Dieni
coniugati (pdf)
Benzene
e aromaticità (pdf)
Legame covalente
Conformazione alcani
Ponte Cloronio
Diels-Alder
Enolo
Mono e Disostituito (pdf)
Lezioni con
MGL Tools / AutoDock
Struttura delle Proteine
Proteine e Legandi
Docking su COX2
Progettare
nuovi farmaci
 Chimica al Computer
Chimica al Computer
PianetaChimica home
|
|
Per seguire questa esercitazione è necessario il programma Arguslab
4.01 che potete scaricare gratuitamente qui:
http://www.arguslab.com
Questa lezione può essere affrontata in due modi:
1) Online. Potete leggere le istruzioni ed eseguirle passo passo
al computer con ArgusLab, aiutandovi con le illustrazioni che chiariscono
ogni passaggio.
2) In aula informatica con la classe. Se siete insegnanti di
chimica, potete adattare la lezione alle esigenze della vostra classe
e proporla in aula informatica ai vostri allievi.
La durata della lezione è di circa due ore.
N.B. Cliccando sulle immagini potrete vederle a pieno schermo.

Gli argomenti di questa prima lezione sono:
-- creazione del modello molecolare di 1-butene, trans-2-butene e cis-2-butene
-- discussione della loro stabilità relativa
-- discussione della maggiore stabilità degli alcheni più
sostituiti attraverso l'esame degli orbitali molecolari HOMO
|
|
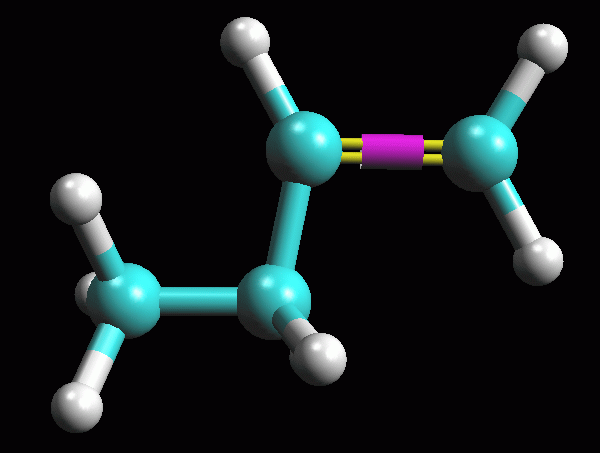
fig. 1
|
|
Creare la molecola di 1-butene
Appena lanciato il programma scegliete file
- new per aprire un nuovo spazio di lavoro. Sono già selezionati
i due pulsanti con la matita ed il programma è pronto per costruire
la molecola. E' già selezionato anche il pulsante C (carbonio)
e la struttura tetraedrica.
-- Generate la catena di carboni. Con un clic del pulsante destro
del mouse (Click destro) generate un carbonio tetraedrico Csp3
che appare giallo (selezionato). Allontanatevi di 1.4 Angstrom e premete
Shift (Maiuscolo) e Click destro. Generate così un
nuovo C legato al precedente. Ripetete l'operazione fino ad avere 4 atomi
di carbonio disposti a zig zag. Uscite dalla modalità aggiungi
atomi cliccando sul pulsante con la freccia gialla  . .
-- Correggete il tipo di atomi. Per trasformare i due carboni C1
e C2 da sp3 a sp2 procedete così: fate Click destro sull'atomo
e scegliete nel menù a finestra Change Atom e poi C[sp2]
- C_2 trigonal non aromatic.
-- Aggiungete gli idrogeni. Cliccate sul pulsante H
in alto  . .
-- Evidenzate il doppio legame. Fate Click destro sul legame
C1-C2 e scegliete Double (fig 1)
Fin qui avete ottenuto una struttura approssimativa di1-butene
come in figura.
|
|
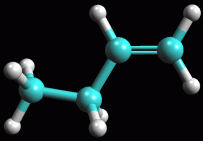
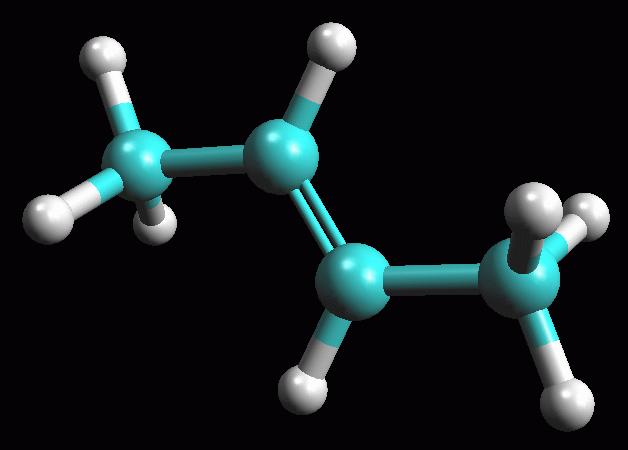
fig. 2
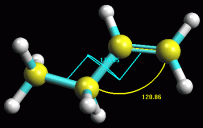
fig. 3
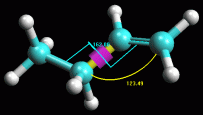
fig. 4
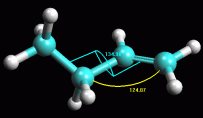
fig. 5

fig. 6
|
|
Ottimizzare la molecola di 1-butene
Per ottimizzare la molecola di 1-butene appena creata, procedete in
due tappe prima usate il metodo UFF (geometrico), poi il medodo AM1
(quantomeccanico).
-- Ottimizzate la geometria col metodo UFF. Nel menù Calculation
scegliete Optimize Geometry e poi nel riquadro Hamiltonian, MM
(Meccanica Molecolare) scegliete UFF (Universal Field Force,
metodo semplice e veloce) e quindi premete OK. Il programma vi
chiederà di salvare prima il file.
-- Salvate. Create una nuova cartella Butene e in questa
salvate la molecola col nome 1-butene.
Solo ora la struttura verrà ottimizzata. Assicuratevi che il
programma scriva Geometry optimization converged!! in caso contrario
ripetete il calcolo più volte premendo il pulsante bunsen  fino a quando ArgusLab otterrà la molecola ottimizzata (fig
2).
fino a quando ArgusLab otterrà la molecola ottimizzata (fig
2).
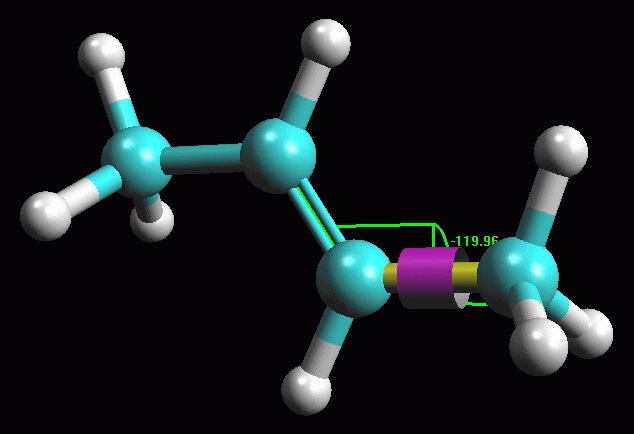
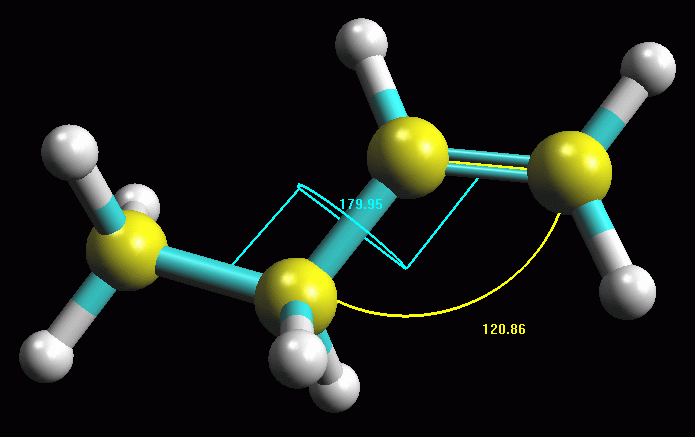
-- Valutate l'angolo di legame e l'angolo diedro-- (Facoltativo):
Fate Ctrl - Click in sequenza sui carboni 1, 2 e 3. Questi diventeranno
gialli, cioè selezionati, e inoltre si evidenzierà il
pulsante in alto a destra con tre atomi azzurri legati  ,
cliccatelo e comparirà un arco con l'indicazione dei gradi dell'angolo
in questione. Evidenziate i carboni 1, 2, 3 e 4, si evidenzierà
il pulsante in alto a destra con 4 atomi azzurri legati ,
cliccatelo e comparirà un arco con l'indicazione dei gradi dell'angolo
in questione. Evidenziate i carboni 1, 2, 3 e 4, si evidenzierà
il pulsante in alto a destra con 4 atomi azzurri legati  ,
cliccatelo e comparirà l'indicazione dei gradi dell'angolo diedro.
in fig 3. Notate che gli angoli indicano notevole simmetria nella
molecola (120 e 180 gradi). ,
cliccatelo e comparirà l'indicazione dei gradi dell'angolo diedro.
in fig 3. Notate che gli angoli indicano notevole simmetria nella
molecola (120 e 180 gradi).
-- Ottimizzate la geometria col metodo AM1. Per questa seconda
ottimizzazione utilizzate una tecnica più sofisticata che tiene
conto del potenziale elettrostatico degli elettroni di valenza, una
tecnica quantomeccanica chiamata semiempirica.
Nel menù Calculation scegliete Optimize Geometry
e poi nel riquadro Hamiltonian, QM (quantomeccanica) scegliete AM1
(Austin Model 1) e quindi premete OK. Per cominciare il calcolo
premete il tasto bunsen. Notate come cambiano gli angoli di legame,
ora non sono più simmetrici, tutta la molecola è stata
ottimizzata in modo raffinato. Ripetete il calcolo (bunsen). L'angolo
sp2 è diventato di 123,5 °, l'angolo
diedro è rimasto 180°.
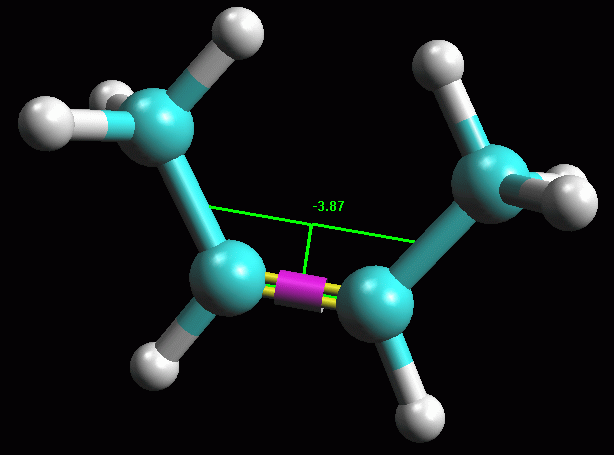
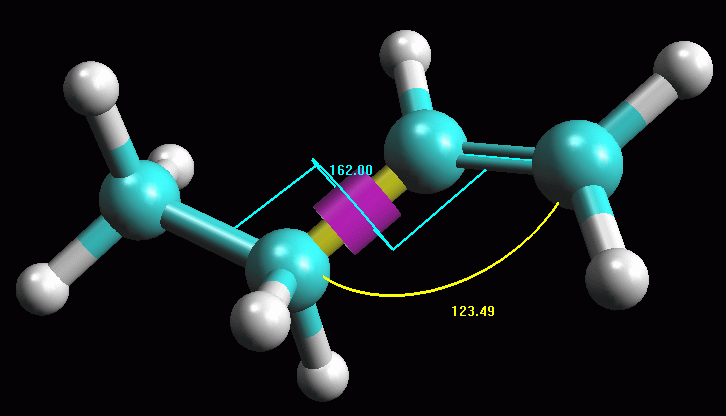
-- Ruotate la molecola attorno al legame C2-C3. Per essere
sicuri che la molecola ottenuta abbia la conformazione più stabile,
provate a perturbarla ruotando l'angolo C2-C3 di qualche grado. Se la
molecola era in un minimo di energia stabile, ottimizzandola ancora
tornerà alla conformazione precedente, altrimenti si porterà
su un nuovo minimo. Fate Click sul legame C2-C3, diventa giallo
con un anello viola. Premete Alt - Shift e trascinate in sù
il mouse col pulsante sinistro premuto. La parte destra della
molecola comincia a ruotare (fig 4).
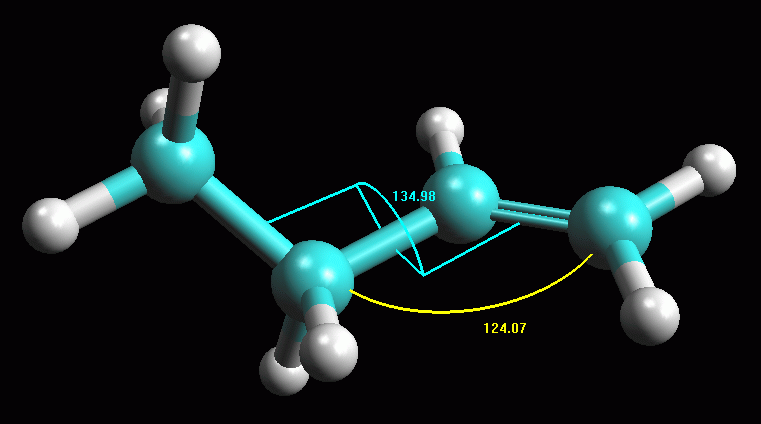
-- Ottimizzate ancora la geometria con AM1. Ripetete l'ottimizzazione
più volte. Noterete che la molecola ruota per portare il gruppo
metile fuori dal piano dell'alchene. La rotazione si arresta quando
l'angolo diedro è intorno ai 135 gradi. Otterrete la molecola
di fig 5.
-- Rimuovete l'indicazione degli angoli di
legame. Nel menù Monitor
scegliete remove all monitors per togliere l'indicazione degli
angoli.
-- Salvate col nome 1-butene_AM1.
Calcolare l'energia di 1-butene
-- Calcolate l'energia. Nel menù Calculation scegliete
Energy, AM1, e quindi premete start.
-- Annotate l'energia. Cliccate il pulsante foglio  dove vedrete i risultati dei calcoli eseguiti. Scorrete fino in fondo
il documento dove troverete il dato sul DH
di formazione della molecola (dovrebbe essere circa DH
= 0,149 kcal/mol)
(fig 6). Copiate questo dato su Word col metodo copia - incolla
(Ctrl-c, Ctrl-v) vi sarà utile per confrontare la stabilità
di questa molecola con quella del trans-2-butene che state per creare.
dove vedrete i risultati dei calcoli eseguiti. Scorrete fino in fondo
il documento dove troverete il dato sul DH
di formazione della molecola (dovrebbe essere circa DH
= 0,149 kcal/mol)
(fig 6). Copiate questo dato su Word col metodo copia - incolla
(Ctrl-c, Ctrl-v) vi sarà utile per confrontare la stabilità
di questa molecola con quella del trans-2-butene che state per creare.
|
|
|
|
|
Creare la molecola di trans-2-butene
-- Trasformate 1-butene in trans-2-butene.
Dal terzo carbonio C3 cancellate un idrogeno: Click (diventa giallo)
e poi premete Canc sulla tastiera. Cambiate il carbonio C3 in C[sp2],
cambiate anche il C1 in C[sp3] (Click
destro, Change Atom).
-- Evidenziate il doppio legame. Ora con Click destro sui legami
scegliete Single sul legame C1-C2 e Double sul legame C2-C3.
-- Aggiungete gli idrogeni cliccando sul pulsante H in alto.
-- Ottimizzate la molecola col metodo UFF (fig. 7)
e poi col metodo AM1 (più volte). Vedrete prima la molecola
ritrovare la geometria planare sui due carboni centrali sp2
coinvolti nel doppio legame, poi vedrete altri aggiustamenti fini che
coinvolgono l'accorciamento del doppio legame e la rotazione dei metili
per portarli in posizione sfalsata. Se i metili non ruotano da soli verso
la posizione sfalsata anti, dovete ruotarli a mano, infatti AM1 si può
fermare anche su un minimo relativo di energia (conformazione eclissata).
Solo dopo che avrete ruotato a mano la molecola, AM1 può trovare
il minimo assoluto della conformazione sfalsata anti.
-- Ruotate i metili. Fate Click sul legame C3-C4 che vene
circondato da un anello viola. Ruotate il metile C4 tenendo premuti
i tasti Alt-Shift e trascinando il mouse in su o in giù
col pulsante sinistro premuto fino a quando l'angolo è di 120 gradi.
(fig. 8). Ripetete la procedura per ruotare anche il metile C1.
-- Ripetete l'ottimizzazione della geometria molecolare con AM1.
-- Salvate la molecola col nome trans-2-butene_AM1. (fig.
9)
|
|

fig.10
|
|
Calcolare l'energia di trans-2-butene
-- Calcolate l'energia della molecola (Calculation,
Energy, AM1) .
-- Annotate l'energia. Cercate in fondo al foglio dei calcoli
il DH di formazione
del 2 butene e copiatelo su Word (dovreste trovare circa -3,36 kcal/mol).
Se trovate un valore più alto (minore in valore assoluto) allora
probabilmente la posizione dei metili non è quella ideale, sfalsata
anti. Ricontrollate l'angolo e ruotate i metili fino a quando l'angolo
indicato è di 120 gradi. A questo punto ripetete l'ottimizzazione
della geometria molecolare con AM1 e ricalcolate l'energia della molecola.
Dovrebbe essere DH
= -3,36 kcal/mol.
(fig. 10)
-- Calcolate la differenza tra le entalpie di formazione di 1-butene
e trans-2-btuene. Dovreste trovare
0,149 + 3,36 = 3,51 kcal/mol (il valore sperimentale è invece
12,5 kcal/mol).
-- Calcolate la K di equilibrio. Da questa differenza di entalpia
DH siete in grado
di calcolare la K di equilibrio di una ipotetica reazione di interconversione
delle due molecole.
2-butene => 1-butene .....................
DH = 3,51 kcal/mol
La relazione da applicare è:
DG° = - RT lnK
quindi
lnK = - DG° / RT
dove l'energia libera G deve essere in J/mol (quindi il DE
in kcal/mol va moltiplicato per 4184),
la costante dei gas R vale 8,314 J/(mol K),
la temperatura T vale 298 K.
lnK = - 3,51 .
4184 / (8,31 .
298) = - 5,93
Approssimando DH a DG,
quindi trascurando l'entropia, dovreste ottenere K = 2,66 10-3.
Da questo si deduce che il trans-2-butene è molto più
stabile di 1-butene come infatti si può verificare nelle reazioni
di eliminazione nelle quali vale la regola di Saytzev: quando
una molecola dà una reazione di eliminazione E1 o E2, si forma
sempre l'alchene più sostituito.
|
|
|
|
|
Creare cis-2-butene e calcolarne l'energia
Trasformate il trans-2-butene in cis-2-butene.
-- Ruotate la molecola rispetto al doppio legame. Fate Click
sul doppio legame centrale e ruotate la parte destra della molecola
fino ad ottenere l'isomero cis, (Alt-Shift e trascinate in giù
il mouse col pulsante sinistro premuto) come in fig 11, l'angolo
indicato diventa zero.
-- Ottimizzate la geometria del cis-2-butene col metodo AM1,
controllate lo stato sfalsato anti dei metili.
-- Calcolate l'energia della molecola col metodo AM1 e
trascrivete il valore dell'entalpia di formazione su Word. Dovreste
trovare circa DH
= -2,22 kcal/mol.
-- Salvate la molecola col nome cis-2-butene_AM1.
Considerate ora un'ipotetica reazione nella quale i due isomeri cis
e trans 2-butene siano in equilibrio.
-- Calcolate la differenza di entalpia tra i due isomeri cis
e trans. Dovreste ottenere che l'isomero trans ha un'energia più
bassa di circa 1,14 kcal/mol (il valore sperimentale è invece
3,5)
cis-2-butene => trans-2-butene ......................
DH = (-3,36 + 2,22) = -1,14 kcal/mol
-- Calcolate la K di equilibrio per dedurre quale potrebbe essere
l'abbondanza relativa dei due isomeri.
lnK = - DG° / RT
lnK = 1,14 .
4184 / (8,31 .
298) = 1,93
Da qui si può ricavare la K di equilibrio. Dovreste trovare K
= 6,89.
Creare gli orbitali molecolari di 1-butene
Tornate alla molecola 1-butene salvata prima 1-butene_AM1 (file,
open).
-- Calcolate gli orbitali. Scegliete Calculation, Energy.
Nella finestra Calculate Properties cliccate Surface Properties
(fig 12). Nella nuova finestra scegliete gli orbitali che volete
esaminare, per esempio 12 Homo, 13 Lumo (fig 13).
Premete OK e nella finestra successiva premete Start.
Il programma calcola la forma e l'energia degli orbitali molecolari
richiesti.
-- Create gli orbitali. Scegliete Surfaces, Make Surfaces,
fate un clic sul + accanto a RHF MOs, vedrete i due file
degli orbitali che avete calcolato, trascinate il primo (12) nella finestra
centrale Grid, scrivete 12 Homo nella finestra subito
sotto e poi cliccate Create>>. Compare il simbolo della
superfice 12 Homo nella finestra di destra. (fig 14).
Ripetete la stessa operazione per l'orbitale 13 Lumo.
|
|
|
|
|
Esaminare l'orbitale Homo di 1-butene
(Highest Occupied Molecular Orbital)
Questo è l'orbitale molecolare occupato a più alta energia
e quindi contiene gli elettroni più reattivi. Quando un elettrofilo
come H+ attacca la molecola, reagisce con gli
elettroni dell'orbitale HOMO. La struttura dell'orbitale HOMO può
aiutare a capire la reattività della molecola verso gli elettrofili.
-- Rappresentate l'orbitale 12 Homo. Scegliete Surfaces,
Make Surfaces, accendete l'icona 12 Homo con il pulsante Toggle
Display (fig 14)e poi date OK. Vedrete l'immagine di
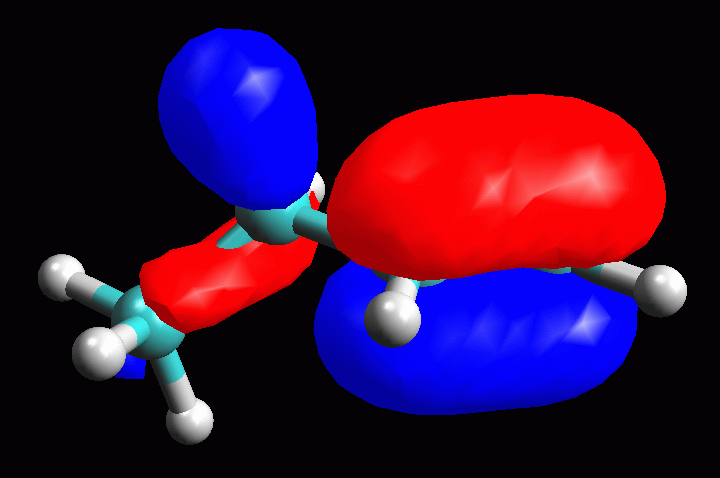
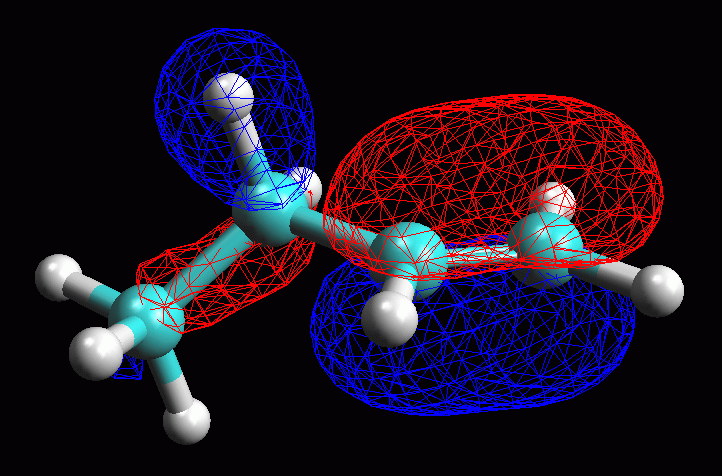
fig 15. L'orbitale è di tipo pigreco con il lobo maggiore
localizzato sul doppio legame, ma con un lobo minore localizzato anche
sui legami CH e CC del carbonio adiacente al doppio legame
-- Scegliere la rappresentazione. Per vedere la struttura della
molecola nascosta dall'orbitale, fate Click destro sulla superficie
dell'orbitale e nel menù che compare scegliete Mesh. (fig
16)
Esaminare l'orbitale Homo del trans 2-butene
Tornate alla molecola di trans-2-butene salvata prima trans-2-butene_AM1
(file, open).
-- Calcolate l'orbitale 12 Homo. Scegliete Calculation, Energy.
Nella finestra Calculate Properties cliccate Surface Properties.
Nella nuova finestra scegliete l'orbitale 12 Homo. Premete OK
e nella finestra successiva premete Start.
-- Create l'orbitale 12 Homo. Scegliete Surfaces, Make
Surfaces, trascinate l'orbitale 12 nella finestra centrale Grid,
scrivete 12 Homo nella finestra subito sotto e poi cliccate Create>>.
Compare una nuova icona nella finestra di destra. Accendete l'icona 12
Homo con il pulsante Toggle Display e date OK. Vedrete l'immagine
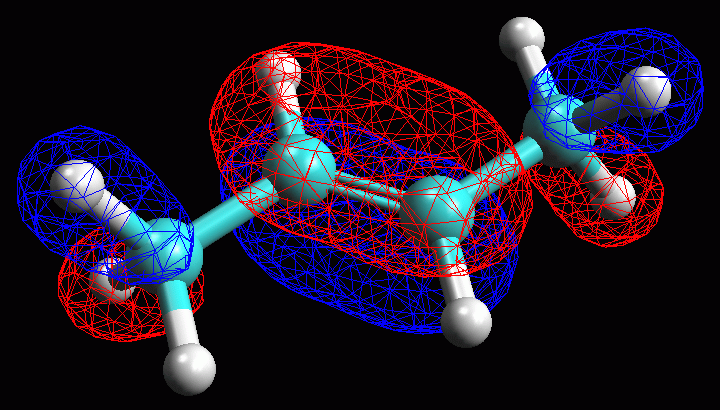
di fig 17. Se scegliete l'opzione Mesh otterrete l'immagine
di fig 18.
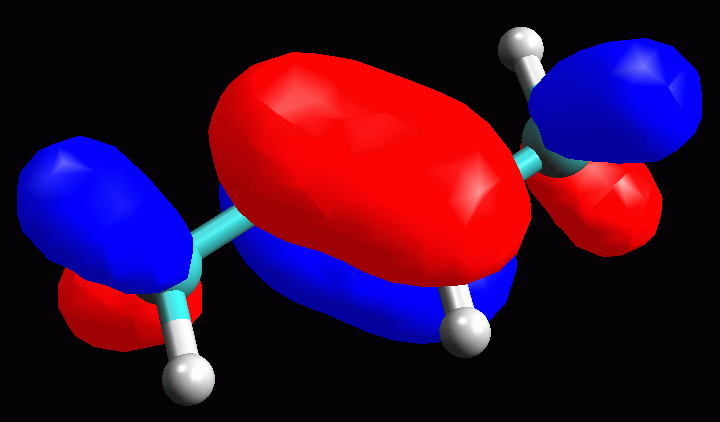
L'orbitale 12 Homo ha il lobo maggiore localizzato sul doppio legame C2-C3,
ma si estende anche ai legami CH adiacenti sul C1 e sul C4. |
|
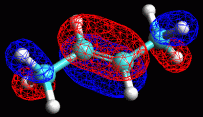
fig. 18
|
|
Gli alcheni più sostituiti sono più stabili
Più sopra abbiamo verificato il dato di fatto sperimentale che
gli alcheni più sostituiti sono più stabili calcolando
con ArgusLab la stabilità di 1-butene e trans-2-butene e trovando
che il trans-2-butene è più stabile di 3,51 kcal/mol (il
valore sperimentale è di 12,5 kcal/mol).
La teoria dice che l'alchene più sostituito è più
stabile a causa dell'effetto induttivo elettron-donatore dei sostituenti
alchilici.
L'effetto induttivo, però, può spiegare soprattutto la
destabilizzazione degli alcheni provocata da sostituenti elettron-attrattori
come Cl e O che, rendendo più positivi i carboni
del doppio legame, fanno diminuire le dimensioni degli orbitali 2p p
e quindi rendono ancora più scarsa la loro sovrapposizione e
più debole il doppio legame. Si pensi, ad esempio alla instabilita
degli enoli.
Con sostituenti alchilici come il metile CH3,
l'effetto induttivo appare insufficiente per spiegare la stabilizzazione
della molecola. D'altra parte il calcolo delle energie con ArgusLab
dimostra che nella struttura di un alchene più sostituito c'è
qualcosa che stabilizza la molecola.
L'osservazione degli orbitali 12 Homo di 1-butene e 2-butene
(gli orbitali con gli elettroni a più alta energia) può
aiutarci a dare una spiegazione qualitativa e intuitiva al problema.
Tra i due alcheni, quello che ha gli elettroni nell'orbitale Homo
più stabilizzati è logico che sia il più
stabile.
L'orbitale Homo ospita gli elettroni p
del doppio legame.
Nell'1-butene (fig 15 e 16) gli elettroni possono occupare
oltre al normale orbitale p
del doppio legame anche uno spazio extra sugli orbitali sp3
del carbonio adiacente e quindi sono stabilizzati.
Nel 2-butene (fig 17 e 18) questa extra stabilizzazione
risulta doppia perchè sono due i carboni vicini
al doppio legame.
Notate che i due idrogeni sui carboni adiacenti al doppio legame sono
disposti uno sopra e uno sotto il piano per poter interagire con i due
lobi (positivo e negativo) dell'orbitale pigreco. Questa interazione
non è così grande da bloccare la rotazione dei metili,
così tutti e tre gli idrogeni sono coinvolti in questo legame
in modo simile.
Anche la teoria VB giunge a conclusioni simili con la sua teoria della
ipeconiugazione che spiega la stabilità del doppio legame coinvolgendo
i legami CH adiacenti.
Autore: prof. Mauro Tonellato
|
|
|
|
|
Chimica al Computer
PianetaChimica home
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|